Appunti di CHIRURGIA PEDIATRICA
Thank you for visiting us
indice
dott. mario leo brena
Il termine ATRESIA CONGENITA DELL'ESOFAGO descrive un folto gruppo di malformazioni che condividono un difetto di continuità esofagea con o senza fistola alla trachea o ai bronchi.
E' una malformazione incompatibile con la vita e la sopravvivenza e la qualità di vita dipende dalla precocità della diagnosi e da un'appropriata terapia chirurgica.
L'incidenza di una atresia esofagea, con o senza fistola, è di circa 1 su 3.000 - 4.500 nati, con una lieve preponderanza per il sesso maschile (rapporto M/F di 3:2).
La maggior parte dei casi si verificano sporadicamente.
Un certo numero di fattori di rischio come i teratogeni ambientali sono ancora discussi in letteratura compresa l'esposizione a talidomide, pillole anticoncezionali, ormoni, e le malattie del sistema endocrino della madre. Inoltre vi sono ampie prove che l'anomalia può essere geneticamente determinata. In primo luogo anomalie cromosomiche si verificano in 6-10% di tutti i casi compresi Trisomia 13 e 18. In secondo luogo un gran numero di sindromi diverse sono state segnalate in associazione con atresia esofagea, ed in ultimo il rischio di ricorrenza di un secondo figlio con la stessa patologia è di circa 0,5-2,0%, mentre il rischio di un neonato nato da un genitore operato per atresia esofagea è di circa il 3,0-4,0%.
La più probabile patogenesi è comunque eterogenea e multifattoriale coinvolgendo cioè diversi geni.
L'embriologia del normale sviluppo dell'intestino è ancora controversa.
Sicuramente il danno avveine precocemente nell'embriogenesi di questa malformazione ed quindi è logico che i bambini con atresia esofagea soffrano di un alto numero di malformazioni associate (50-80%).
Le anomalie associate più frequenti sono le malformazioni muscoloscheletriche (20-70%), seguite da quelle cardiovascolari (20-50%), genito-urinario (15-25%), gastrointestinale (15-25%) e anomalie cromosomiche (5-10%).
Le anomalia cardiache, che sono poi le vere responsabili della mortalità in questa malforamzione, sono quelle del difetto del setto ventricolare (19%), del setto interatriale (20%), la tetralogia di Fallot (5%), la coartazione aortica (1%) o l'arco aortico destro (4%). E 'importante rendersi conto che alcuni di questi difetti cardiaci possono portare ad una insufficienza cardiaca clinicamente evidente in pochi giorni dal parto. Pertanto, tutti i pazienti con atresia esofagea dovrebbere essere subito sottoposti ad indagine ecocardiografica nonché ad esame ecografica renale e dell'encefalo.
La più comune anomalia gastrointestinale associata è l'atresia ano-rettale (9%) seguita dall'atresia duodenale (5%), malrotazione (4%) e da altre atresie intestinali (1%).
Nel 10% dei casi i pazienti affetti da atresia esofagea possono essere fatti rientrare in specifiche sindromi. Fra queste per esempio la sindrome di Holt, di Oram, di DiGeorge, di Goldenhair, le trisomie 13-18-21, l'associazione CHARGE (coloboma, difetti cardiaci, atresia delle coane, ritardo mentale, anomalie dei genitali, malformazioni dell'orecchio) e molte altre.
Il modello di comuni anomalie associate ha portato alla creazione dell'associazione indicata con l'acronimo VATER (anomalie vertebrali, atresia anale, fistola tracheo-esofagea, anomalie renali) o VACTERL (in aggiunta: anomalie cardiache e degli arti). L'incidenza di queste associazioni è circa del 20% nella popolazione affetta da atresia esofagea, ma due o più anomalie si verificano in quasi la metà dei pazienti.
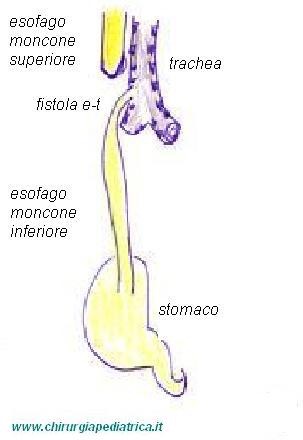
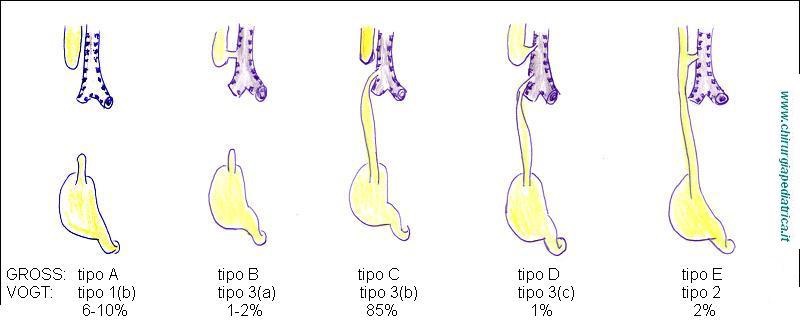
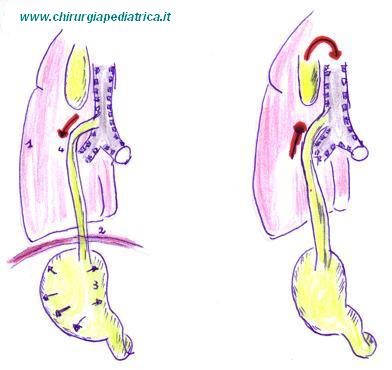
Le varie classificazioni della malformazione di solito prendono il loro orientamento dalla presenza e tipo di fistola tracheo-esofagea.
Dal punto di vista clinico si definiscono:
tipo 1: atresia senza fistola esofago-tracheale (8,5%)
tipo 2: atresia con fistola esofago-tracheale al moncone superiore (1%)
tipo 3: atresia con fistola esofago-tracheale al moncone inferiore (85%)
tipo 4: atresia con fistola esofago-tracheale su entrambi i monconi (1,5%)
tipo 5: solo fistola esofago-tracheale (4%)
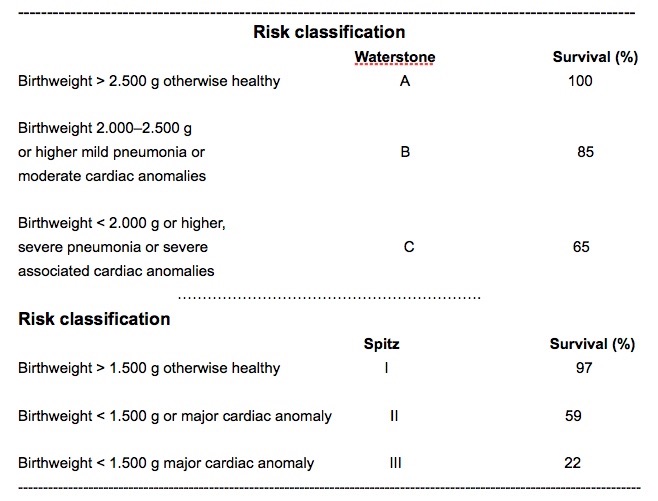
In aggiunta a queta classificazione vi sono poi quelle legate al rischio di mortalità basate sul peso alla nascita, anomalie cardiache, patologie polmonari, che permettono di confrontare i risultati delle diverse casisitiche. In particolare le malformazioni cardiache sono quelle che hanno un maggior significato sul tasso di mortalità in questi piccoli pazienti (vedi scheda accanto).
Un segno indicativo di sospetta atresia dell'esofago in corso di gravidanza è il polidramnios. Polidramnios è una manifestazione aspecifica di un disturbo della deglutizione o della mancata progresione del contenuto intestinale nel feto. L'ecografia prenatale può inoltre rivelare uno spostamento in avanti ed indietro dei fluidi nella tasca esofagea superiore e, nei casi di assenza della fistola, una scarsità di fluido in stomaco e intestino tenue.
La presentazione postnatale della malformazione è caratterizzato da drooling di saliva, soffocamento, tosse ed episodi di cianosi. Una prova di alimentazione è controindicata con questi sintomi perché è la causa prima di aspirazione e polmonite.
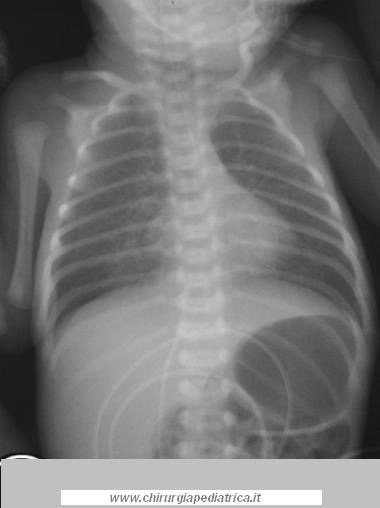
Il sucessivo step diagnostico è quello di posizionare un sondino naso-gastrico. Se questo non è possibile con arresto della progressione della sonda a circa 10-12 cm dall'ADS, la diagnosi di atresia esofagea è quasi certa. Per questo motivo sondini di piccolo calibro devono essere evitati in quanto arrotolandosi nella tasca superiore possono dare l'illusione di essersi spinti fin nello stomaco. L'esame obiettivo deve essere completato con la ricerca di eventuali malformazioni associate.
Il passo successivo è quello di eseguire una semplice radiografia comprensiva di collo, torace e addome. L'approssimativa lunghezza del pauch superiore può essere stimato dalla lunghezza della sonda inserita. Aria al di sotto del diaframma può essere vista in presenza di una fistola tracheoesofagea distale e ulteriori livelli idroaerei possono indicare un'atresia duodenale o intestinale. Un addome privo di gas indica un'atresia esofagea senza fistola al moncono inferiore. La traslucenza dei polmoni fornisce le informazioni su una possibile polmonite ab ingestis.
La valutazione cardiologica comprende un'ecocardiografia di routine per valutare le malformazioni associate e la posizione dell'arco aortico, importante per valutare l'accesso chirurgico.
Le altre indagini prevedono l'ecografia addominale e lo studio di eventuali aomalie schelettriche della colonna vertebrale o delle coste.
I bambini sono allettati in terapia intensiva neonatale. L' intervento chirurgico immediato è raramente richiesto; di conseguenza, tutte le suddette indagini possono essere eseguite passo per passo. L'inserimento di un tubo oro- o nasoesofageo tipo Replongle doppio lume è obbligatorio per l'aspirazione continua o intermittente della saliva al fine di prevenirne l'inalazione.
Il bambino deve essere posti in posizione anti-trendelemburg per ridurre al minimo il reflusso gastroesofageo nella trachea e nei polmoni attraverso la fistola inferiore.
L'intubazione e la ventilazione è necessaria solo in caso di difficoltà respiratoria, polmonite grave, o altre malformazioni che compromettono la respirazione spontanea.
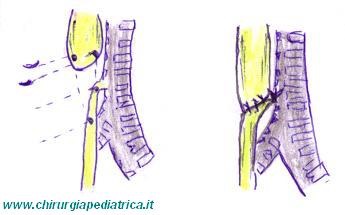
L'obiettivo della correzione chirurgica è quello di dividere la fistola (se presente) per chiuderla sul lato della trachea ed eseguire quindi un'anastomosi fra i due monconi esofagei. L'approccio standard è attraverso una toracotomia dorsale laterale destra per via extrapleurica attraverso il quarto spazio intercostale.
E' possibile la riparazione dell'atresia per via toracoscopica, ma questa è una procedura di elevata difficoltà e necessita pertando di un'elevata esperienza in chirurgia endoscopica.
Se vi è l'arco aortico a destra vi è indicato un approccio tramite una toracotomia sinistra.
Se disponibile è possibile eseguire una tracheoscopia pre-operatoria per individuare la presenza diuna fistola anche del moncone superiore.
Nella maggior parte dei casi l'anastomosi può essere compiuta senza particolare tensione fra i margini. Se la tensione sembra essere troppa, nonostante la mobilitazione del moncone inferiore e superiore dell'esofago, un ulteriore allungamento può essere ottenuto con una miotomia circolare sec. Livatitis. Tuttavia questa procedura può determinare la formazione di un pseudodiverticolo oltre ad un grave problema di ipomobilità dell'esofago per cui è da molti stata abbandonata. Un altro modo per ridurre la tensione all'anastomosi in alcuni casi è quello eseguire un flap di mucosa e muscolare dal esofageo superiore pouch.
DIverso invece l'atteggiamento in caso di Atresia esofagea isolata (tipo1): The Long Gap Problem (8,5%).
Una radiografia dell'addome priva di gas è sospetta per un'atresia esofagea senza fistola distale. L'anastomosi diretta di solito non è possibile in questi casi per l'enorme distanza tra i monconi esofagei. Pertanto, la procedura primaria consiste nell'eseguire una gastrostomia per consentire l'alimentazione del bambino.
La distanza di tre o quattro corpi vertebrali, valutati all'Rx torace, fra i monconi è considerato come limite per definire un'atresia di tipo long-gap e una distanza di 5 o più corpi vertebrali è considerata una variante ultra-lunga.
Due strategie chirurgiche di base sono disponibili in caso di atresia esofagea long gap. La prima è la conservazione dell'esofago del paziente con ritardo nella ricanalizzazione attendono cioè la crescita spontanea dei monconi, più pronunciata in quello superiore, ma che ha bisogno di 12-16 settimane, in media, prima di poter raggiungere la condizione ottimale per poter eseguire l'anastomosi esofago-esofagea. L'alternativa è la sostituzione esofagea con altro tratto di intestino.
Una modifica alla tecnica di Rehbein che prevede la trazione esterna dei monconi, è stata descritta da Foker.
In ogni caso, è essenziale poter dimostrare che un segmento esofageo inferiore esiste veramente. Se questo non ci fosse la sostituzione esofagea deve essere pianificata come primo approccio. Per la sostituzione esofagea si possono utilizzare tratti di colon, digiuno o tramite un pull-up gastrico.
Nonostante il tasso di sopravvivenza a lungo termine sia eccellente, ci sono un gran numero di complicanze precoci e tardive che hanno bisogno di speciali cure e attenzione.
L'incidenza di complicanze precoci è stata ridotta in modo significativo negli ultimi decenni. Oggi, un neonato con atresia esofagea viene ricosciuto subito alla nascita evitando le complicanze legate all'inalazione. Inoltre le tecniche chirurgiche sono state affinate come pure il materiale di sutura. Infine, i progressi nell'assistenza post-operatoria, contribuiscono in larga misura a garantire un'ottima sopravvivenza per i pazienti appartenenti ai gruppi A e B di Waterston.
Una complicazione precoce, ma fortunatamente rara, è la ricorrenza della fistola tracheo-esofagea (3%). La spontaneo chiusura della stessa è rara e quindi necessita di un nuovo intervento. Questa procedura è resa difficile dal locale processo infiammatorio ed edema con peggioramento della consistenza dei tessuti su cui si va a comporre la sutura.
L'incidenza di un leak anastomotico è in genere inferiore al 10% dei casi se la tensione dell'anastomosi non è ecessiva. Nella maggior parte di questi casi i difetti sono piccoli e clinicamente insignificanti ed il trattamento è in genere conservativo.
Eseguiamo un esofagogramma di controllo nelle prime settimane dopo l'intervento proprio per valutare sia la presenza della fistola che della stenosi dell'anastomosi.
Se le condizioni del paziente sono stabili l'alimentazione per via orale è iniziata precocemente.
La stenosi anastomotica (vedi immagine a lato) è una complicanza molto comune. L'esofagogramma mostra spesso un anastomosi stretta anche a causa delle differenze di calibro dei segmenti esofageo accostati. In quest'ultimo caso l'alimentazione è sempre ben tollerata. Una vera stenosi cicatriziale non migliora spontaneamente e provoca disturbi nell'alimentazione.
Se la stenosi è minima può essere trattata con successo tramite una serie di dilatazioni esofagee per via endoscopica. Una complicanza di questa manovra è la rottura dell'esofago. Il mantenimento cronico della stenosi è in genere determinato dalla concomitante presenza di un reflusso gastro-esofageo per cui diviene determinante la corretta terapia con inibitori di pompa anche per un lungo periodo. Possibile la necessità di ricorrere ad una fundoplicatio se non si hanno miglioramenti.
La Tracheomalacia è comunemente associata nei pazienti affetti da atresia dell'esofago. Essa causa il collasso della trachea determinando la tipica tosse abbaiante ed uno stridore inspiratorio. Nella maggior parte dei casi risulta essere però autolimitante con risoluzione spontanea nei primi mesi/anno di vita. Tuttavia le forme più gravi di tracheomalacia possono portare ad insufficienza respiratoria, difficoltà nell'alimentazione e gravi crisi d'apnea. In questi casi può rendersi necessario intervento di aortopexy o in alternativa con il posizionamento di una stent per via endoscopica con risultati però alterni.
La complicanza più comune è il reflusso gastroesofageo (vedi argomento a parte) che può dare problemi di alimentazione, vomito, scarso accrescimento e ricorrenti infezioni del tratto respiratorio.
Il reflusso è favorito dalla trazione sul moncone esofageo inferiore che si deve compiere per poter eseguire l'anastomosi. In contrasto con i bambini altrimenti normali, non c'è quindi la possibilità di maturazione spontanea dei disturbi esofagei. L'esposizione cronica dell'anastomosi al reflusso acido è una causa ben nota di stenosi recidiva o di esofago di Barrett.
Inoltre, la peristalsi propulsiva nel segmento inferiore dell'esofago manca e il tempo di clearance del materiale acido è significativamente più lungo rispetto al normale.
Quindi molti dei pazienti con atresia esofagea hanno alla fine bisogno di una fundoplicatio.
Comuni problemi a lungo termine sono poi le ricorrenti infezioni delle vie respiratorie a causa di microaspirazioni per qualche tipo di disturbo della deglutizione o a causa della presenza di un'anomala attività peristaltica nel segmento inferiore.
In conclusione, i neonati con atresia esofagea hanno una prognosi eccellente se non sono presenti altre gravi malformazioni.
Tuttavia i problemi continuano ad esistere per i neonati con atresia esofage di tipo long-gap. In quest'ultimo caso il mantenere l'esofago nativo del paziente stesso è pagato però a volte con gravi problemi a lungo termine a causa delle anomale della peristalsi, delle ricorrenti stenosi e del grave reflusso gastroesofageo. L'alternativa sostituzione dell'oragano con altro intestino è però altrettanto problematica per cui non si è ancora individuato un metodo ideale per affrontare la variante long-gap di questa patologia.
E' una malformazione incompatibile con la vita e la sopravvivenza e la qualità di vita dipende dalla precocità della diagnosi e da un'appropriata terapia chirurgica.
L'incidenza di una atresia esofagea, con o senza fistola, è di circa 1 su 3.000 - 4.500 nati, con una lieve preponderanza per il sesso maschile (rapporto M/F di 3:2).
La maggior parte dei casi si verificano sporadicamente.
Un certo numero di fattori di rischio come i teratogeni ambientali sono ancora discussi in letteratura compresa l'esposizione a talidomide, pillole anticoncezionali, ormoni, e le malattie del sistema endocrino della madre. Inoltre vi sono ampie prove che l'anomalia può essere geneticamente determinata. In primo luogo anomalie cromosomiche si verificano in 6-10% di tutti i casi compresi Trisomia 13 e 18. In secondo luogo un gran numero di sindromi diverse sono state segnalate in associazione con atresia esofagea, ed in ultimo il rischio di ricorrenza di un secondo figlio con la stessa patologia è di circa 0,5-2,0%, mentre il rischio di un neonato nato da un genitore operato per atresia esofagea è di circa il 3,0-4,0%.
La più probabile patogenesi è comunque eterogenea e multifattoriale coinvolgendo cioè diversi geni.
L'embriologia del normale sviluppo dell'intestino è ancora controversa.
Sicuramente il danno avveine precocemente nell'embriogenesi di questa malformazione ed quindi è logico che i bambini con atresia esofagea soffrano di un alto numero di malformazioni associate (50-80%).
Le anomalie associate più frequenti sono le malformazioni muscoloscheletriche (20-70%), seguite da quelle cardiovascolari (20-50%), genito-urinario (15-25%), gastrointestinale (15-25%) e anomalie cromosomiche (5-10%).
Le anomalia cardiache, che sono poi le vere responsabili della mortalità in questa malforamzione, sono quelle del difetto del setto ventricolare (19%), del setto interatriale (20%), la tetralogia di Fallot (5%), la coartazione aortica (1%) o l'arco aortico destro (4%). E 'importante rendersi conto che alcuni di questi difetti cardiaci possono portare ad una insufficienza cardiaca clinicamente evidente in pochi giorni dal parto. Pertanto, tutti i pazienti con atresia esofagea dovrebbere essere subito sottoposti ad indagine ecocardiografica nonché ad esame ecografica renale e dell'encefalo.
La più comune anomalia gastrointestinale associata è l'atresia ano-rettale (9%) seguita dall'atresia duodenale (5%), malrotazione (4%) e da altre atresie intestinali (1%).
Nel 10% dei casi i pazienti affetti da atresia esofagea possono essere fatti rientrare in specifiche sindromi. Fra queste per esempio la sindrome di Holt, di Oram, di DiGeorge, di Goldenhair, le trisomie 13-18-21, l'associazione CHARGE (coloboma, difetti cardiaci, atresia delle coane, ritardo mentale, anomalie dei genitali, malformazioni dell'orecchio) e molte altre.
Il modello di comuni anomalie associate ha portato alla creazione dell'associazione indicata con l'acronimo VATER (anomalie vertebrali, atresia anale, fistola tracheo-esofagea, anomalie renali) o VACTERL (in aggiunta: anomalie cardiache e degli arti). L'incidenza di queste associazioni è circa del 20% nella popolazione affetta da atresia esofagea, ma due o più anomalie si verificano in quasi la metà dei pazienti.
Le varie classificazioni della malformazione di solito prendono il loro orientamento dalla presenza e tipo di fistola tracheo-esofagea.
Dal punto di vista clinico si definiscono:
tipo 1: atresia senza fistola esofago-tracheale (8,5%)
tipo 2: atresia con fistola esofago-tracheale al moncone superiore (1%)
tipo 3: atresia con fistola esofago-tracheale al moncone inferiore (85%)
tipo 4: atresia con fistola esofago-tracheale su entrambi i monconi (1,5%)
tipo 5: solo fistola esofago-tracheale (4%)
In aggiunta a queta classificazione vi sono poi quelle legate al rischio di mortalità basate sul peso alla nascita, anomalie cardiache, patologie polmonari, che permettono di confrontare i risultati delle diverse casisitiche. In particolare le malformazioni cardiache sono quelle che hanno un maggior significato sul tasso di mortalità in questi piccoli pazienti (vedi scheda accanto).
Un segno indicativo di sospetta atresia dell'esofago in corso di gravidanza è il polidramnios. Polidramnios è una manifestazione aspecifica di un disturbo della deglutizione o della mancata progresione del contenuto intestinale nel feto. L'ecografia prenatale può inoltre rivelare uno spostamento in avanti ed indietro dei fluidi nella tasca esofagea superiore e, nei casi di assenza della fistola, una scarsità di fluido in stomaco e intestino tenue.
La presentazione postnatale della malformazione è caratterizzato da drooling di saliva, soffocamento, tosse ed episodi di cianosi. Una prova di alimentazione è controindicata con questi sintomi perché è la causa prima di aspirazione e polmonite.
Il sucessivo step diagnostico è quello di posizionare un sondino naso-gastrico. Se questo non è possibile con arresto della progressione della sonda a circa 10-12 cm dall'ADS, la diagnosi di atresia esofagea è quasi certa. Per questo motivo sondini di piccolo calibro devono essere evitati in quanto arrotolandosi nella tasca superiore possono dare l'illusione di essersi spinti fin nello stomaco. L'esame obiettivo deve essere completato con la ricerca di eventuali malformazioni associate.
Il passo successivo è quello di eseguire una semplice radiografia comprensiva di collo, torace e addome. L'approssimativa lunghezza del pauch superiore può essere stimato dalla lunghezza della sonda inserita. Aria al di sotto del diaframma può essere vista in presenza di una fistola tracheoesofagea distale e ulteriori livelli idroaerei possono indicare un'atresia duodenale o intestinale. Un addome privo di gas indica un'atresia esofagea senza fistola al moncono inferiore. La traslucenza dei polmoni fornisce le informazioni su una possibile polmonite ab ingestis.
La valutazione cardiologica comprende un'ecocardiografia di routine per valutare le malformazioni associate e la posizione dell'arco aortico, importante per valutare l'accesso chirurgico.
Le altre indagini prevedono l'ecografia addominale e lo studio di eventuali aomalie schelettriche della colonna vertebrale o delle coste.
I bambini sono allettati in terapia intensiva neonatale. L' intervento chirurgico immediato è raramente richiesto; di conseguenza, tutte le suddette indagini possono essere eseguite passo per passo. L'inserimento di un tubo oro- o nasoesofageo tipo Replongle doppio lume è obbligatorio per l'aspirazione continua o intermittente della saliva al fine di prevenirne l'inalazione.
Il bambino deve essere posti in posizione anti-trendelemburg per ridurre al minimo il reflusso gastroesofageo nella trachea e nei polmoni attraverso la fistola inferiore.
L'intubazione e la ventilazione è necessaria solo in caso di difficoltà respiratoria, polmonite grave, o altre malformazioni che compromettono la respirazione spontanea.
L'obiettivo della correzione chirurgica è quello di dividere la fistola (se presente) per chiuderla sul lato della trachea ed eseguire quindi un'anastomosi fra i due monconi esofagei. L'approccio standard è attraverso una toracotomia dorsale laterale destra per via extrapleurica attraverso il quarto spazio intercostale.
E' possibile la riparazione dell'atresia per via toracoscopica, ma questa è una procedura di elevata difficoltà e necessita pertando di un'elevata esperienza in chirurgia endoscopica.
Se vi è l'arco aortico a destra vi è indicato un approccio tramite una toracotomia sinistra.
Se disponibile è possibile eseguire una tracheoscopia pre-operatoria per individuare la presenza diuna fistola anche del moncone superiore.
Nella maggior parte dei casi l'anastomosi può essere compiuta senza particolare tensione fra i margini. Se la tensione sembra essere troppa, nonostante la mobilitazione del moncone inferiore e superiore dell'esofago, un ulteriore allungamento può essere ottenuto con una miotomia circolare sec. Livatitis. Tuttavia questa procedura può determinare la formazione di un pseudodiverticolo oltre ad un grave problema di ipomobilità dell'esofago per cui è da molti stata abbandonata. Un altro modo per ridurre la tensione all'anastomosi in alcuni casi è quello eseguire un flap di mucosa e muscolare dal esofageo superiore pouch.
DIverso invece l'atteggiamento in caso di Atresia esofagea isolata (tipo1): The Long Gap Problem (8,5%).
Una radiografia dell'addome priva di gas è sospetta per un'atresia esofagea senza fistola distale. L'anastomosi diretta di solito non è possibile in questi casi per l'enorme distanza tra i monconi esofagei. Pertanto, la procedura primaria consiste nell'eseguire una gastrostomia per consentire l'alimentazione del bambino.
La distanza di tre o quattro corpi vertebrali, valutati all'Rx torace, fra i monconi è considerato come limite per definire un'atresia di tipo long-gap e una distanza di 5 o più corpi vertebrali è considerata una variante ultra-lunga.
Due strategie chirurgiche di base sono disponibili in caso di atresia esofagea long gap. La prima è la conservazione dell'esofago del paziente con ritardo nella ricanalizzazione attendono cioè la crescita spontanea dei monconi, più pronunciata in quello superiore, ma che ha bisogno di 12-16 settimane, in media, prima di poter raggiungere la condizione ottimale per poter eseguire l'anastomosi esofago-esofagea. L'alternativa è la sostituzione esofagea con altro tratto di intestino.
Una modifica alla tecnica di Rehbein che prevede la trazione esterna dei monconi, è stata descritta da Foker.
In ogni caso, è essenziale poter dimostrare che un segmento esofageo inferiore esiste veramente. Se questo non ci fosse la sostituzione esofagea deve essere pianificata come primo approccio. Per la sostituzione esofagea si possono utilizzare tratti di colon, digiuno o tramite un pull-up gastrico.
Nonostante il tasso di sopravvivenza a lungo termine sia eccellente, ci sono un gran numero di complicanze precoci e tardive che hanno bisogno di speciali cure e attenzione.
L'incidenza di complicanze precoci è stata ridotta in modo significativo negli ultimi decenni. Oggi, un neonato con atresia esofagea viene ricosciuto subito alla nascita evitando le complicanze legate all'inalazione. Inoltre le tecniche chirurgiche sono state affinate come pure il materiale di sutura. Infine, i progressi nell'assistenza post-operatoria, contribuiscono in larga misura a garantire un'ottima sopravvivenza per i pazienti appartenenti ai gruppi A e B di Waterston.
Una complicazione precoce, ma fortunatamente rara, è la ricorrenza della fistola tracheo-esofagea (3%). La spontaneo chiusura della stessa è rara e quindi necessita di un nuovo intervento. Questa procedura è resa difficile dal locale processo infiammatorio ed edema con peggioramento della consistenza dei tessuti su cui si va a comporre la sutura.
L'incidenza di un leak anastomotico è in genere inferiore al 10% dei casi se la tensione dell'anastomosi non è ecessiva. Nella maggior parte di questi casi i difetti sono piccoli e clinicamente insignificanti ed il trattamento è in genere conservativo.
Eseguiamo un esofagogramma di controllo nelle prime settimane dopo l'intervento proprio per valutare sia la presenza della fistola che della stenosi dell'anastomosi.
Se le condizioni del paziente sono stabili l'alimentazione per via orale è iniziata precocemente.
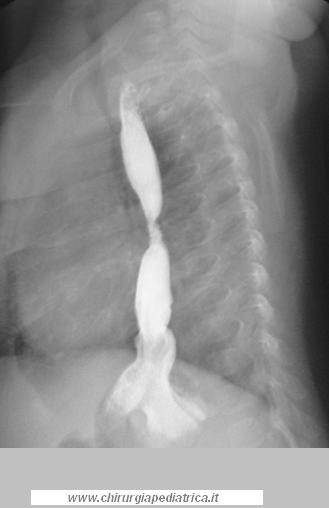
La stenosi anastomotica (vedi immagine a lato) è una complicanza molto comune. L'esofagogramma mostra spesso un anastomosi stretta anche a causa delle differenze di calibro dei segmenti esofageo accostati. In quest'ultimo caso l'alimentazione è sempre ben tollerata. Una vera stenosi cicatriziale non migliora spontaneamente e provoca disturbi nell'alimentazione.
Se la stenosi è minima può essere trattata con successo tramite una serie di dilatazioni esofagee per via endoscopica. Una complicanza di questa manovra è la rottura dell'esofago. Il mantenimento cronico della stenosi è in genere determinato dalla concomitante presenza di un reflusso gastro-esofageo per cui diviene determinante la corretta terapia con inibitori di pompa anche per un lungo periodo. Possibile la necessità di ricorrere ad una fundoplicatio se non si hanno miglioramenti.
La Tracheomalacia è comunemente associata nei pazienti affetti da atresia dell'esofago. Essa causa il collasso della trachea determinando la tipica tosse abbaiante ed uno stridore inspiratorio. Nella maggior parte dei casi risulta essere però autolimitante con risoluzione spontanea nei primi mesi/anno di vita. Tuttavia le forme più gravi di tracheomalacia possono portare ad insufficienza respiratoria, difficoltà nell'alimentazione e gravi crisi d'apnea. In questi casi può rendersi necessario intervento di aortopexy o in alternativa con il posizionamento di una stent per via endoscopica con risultati però alterni.
La complicanza più comune è il reflusso gastroesofageo (vedi argomento a parte) che può dare problemi di alimentazione, vomito, scarso accrescimento e ricorrenti infezioni del tratto respiratorio.
Il reflusso è favorito dalla trazione sul moncone esofageo inferiore che si deve compiere per poter eseguire l'anastomosi. In contrasto con i bambini altrimenti normali, non c'è quindi la possibilità di maturazione spontanea dei disturbi esofagei. L'esposizione cronica dell'anastomosi al reflusso acido è una causa ben nota di stenosi recidiva o di esofago di Barrett.
Inoltre, la peristalsi propulsiva nel segmento inferiore dell'esofago manca e il tempo di clearance del materiale acido è significativamente più lungo rispetto al normale.
Quindi molti dei pazienti con atresia esofagea hanno alla fine bisogno di una fundoplicatio.
Comuni problemi a lungo termine sono poi le ricorrenti infezioni delle vie respiratorie a causa di microaspirazioni per qualche tipo di disturbo della deglutizione o a causa della presenza di un'anomala attività peristaltica nel segmento inferiore.
In conclusione, i neonati con atresia esofagea hanno una prognosi eccellente se non sono presenti altre gravi malformazioni.
Tuttavia i problemi continuano ad esistere per i neonati con atresia esofage di tipo long-gap. In quest'ultimo caso il mantenere l'esofago nativo del paziente stesso è pagato però a volte con gravi problemi a lungo termine a causa delle anomale della peristalsi, delle ricorrenti stenosi e del grave reflusso gastroesofageo. L'alternativa sostituzione dell'oragano con altro intestino è però altrettanto problematica per cui non si è ancora individuato un metodo ideale per affrontare la variante long-gap di questa patologia.
ATRESIA DELL'ESOFAGO